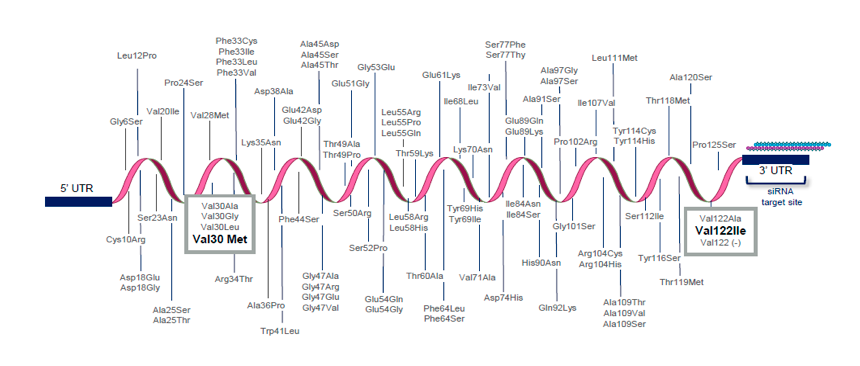
Amiloidosi ATTR ereditaria
La diagnosi della patologia può essere effettuata mediante il test genetico per l’Amiloidosi ATTR ereditaria. Si tratta di un test diagnostico che si esegue su un campione di DNA di tipo germinale e permette di identificare in tempi rapidi la presenza di una mutazione a carico del gene responsabile della sintesi della proteina transtiretina localizzato sul cromosoma 18.1 Rappresenta il principale strumento per formulare una diagnosi quando esiste un sospetto diagnostico.
Porfiria Epatica Acuta
I test diagnostici in grado di confermare la diagnosi di Porfiria Epatica Acuta sono i test biochimici, ma è opportuno eseguire un test genetico per confermare il sottotipo di AHP di cui il paziente è affetto. E' indicato effettuare il test genetico dei membri della famiglia affetti da AHP per identificare i soggetti portatori della mutazione genetica e potenzialmente predisposti per lo sviluppo della sintomatologia.
Iperossaluria Primaria di Tipo 1
La Primary Hyperoxaluria (PH) coinvolge il metabolismo del gliossilato, precursore dell’ossalato. Esistono tre diverse forme di iperossaluria primitiva, il tipo 1 (PH1), il tipo 2 (PH2) e il tipo 3 (PH3), ognuna delle quali caratterizzata da uno specifico difetto enzimatico. La causa di PH1 è una carenza di un enzima specifico per il fegato chiamato alanina-gliossilato aminotransferasi (AGT), derivante da una mutazione del gene AGXT.